Informació clínica
Utilitat diagnòstica:
Identificar el defecte molecular al F8 en pacients diagnosticats d'HA.
Hemofília A (HA)
L'HA és la forma més freqüent d'hemofília amb una prevalença estimada de 1:5.000 homes. Aquest trastorn hemorràgic està causat per una deficiència de factor VIII (FVIII) de la coagulació. Segons els nivells de FVIII es distingeixen tres formes d'HA: severa si l'activitat biològica del FVIII és <1%; moderada si l'activitat del FVIII està entre 1-5%; lleu si l'activitat del FVIII està entre 5-40%. Les manifestacions clíniques més freqüents són les hemorràgies a les articulacions (hemartrosi) i als músculs (hematomes). La freqüència i severitat dels sagnats és inversament proporcional a la quantitat de FVIII que hi ha al plasma.
L'HA presenta una herència recessiva lligada al cromosoma X, de manera que els homes són afectes i les dones són portadores de la patologia. Aquest trastorn està causat per mutacions al gen que codifica per la proteïna FVIII de la coagulació (F8) que poden afectar a l'activitat del FVIII de la coagulació o reduir la quantitat d'aquesta proteïna. Actualment s'han identificat més de 1.300 alteracions genètiques al F8, entre les quals s'inclouen canvis d'un sol nucleòtid i delecions o insercions de varis nucleòtids. No obstant, en els casos d'HA greu la mutació més freqüent és la inversió de l'intró 22, present en un 50% dels pacients. Aquesta mutació succeeix per una recombinació homòloga entre el F8 i l'extrem telomèric del cromosoma X degut a l'alta homologia que existeix entre ambdues seqüències
Aplicació d'un panell de múltiples gens que es basa en l'amplificació simultània dels exons i les regions intròniques flanquejants per a la seva seqüenciació mitjançant tècniques de seqüenciació massiva (NGS) i permet realitzar l'estudi molecular simultani dels gens relacionats amb les coagulopaties congènites i trastorns hemorràgics hereditaris entre els quals es troba el gen del factor VIII (F8).
Mètode:
Seqüenciació massiva (NGS)
Anàlisi de les inversions de l'intró 1 mitjançant PCR convencional i de l'intró 22 mitjançant PCR llarga (LR-PCR) al F8.
Seqüenciació massiva dels exons i les regions intròniques flanquejants del F8 amb l'aplicació d'un panell de gens relacionats amb les coagulopaties congènites.
Seqüenciació tradicional de Sanger per recomprovar la/les mutació/ns detectades en els pacients diagnosticats amb HA, per tal d'arribar a un resultat inequívoc, analitzant la regió concreta on es troba la variant.
En el cas de no identificar cap mutació potencial o definitivament causant de la patologia s'informarà i discutirà amb l'equip mèdic demandant de la prova la possibilitat de realitzar estudis complementaris.
Valors de referència
No aplica
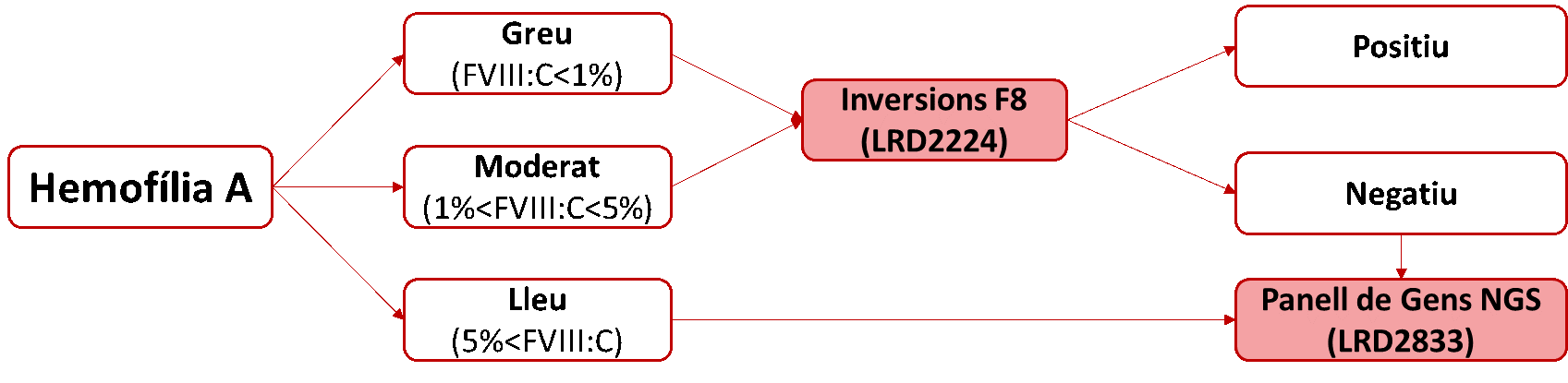
Algoritme diagnòstic:
En funció del fenotip del pacient s'inclou:
- HA greu (FVIII:C<1%) i HA moderat (1%<FVIII:C<5%): Estudi de les inversions de l'intró 22 i de l'intró 1.
- En el cas de ser negatives i en pacients amb diagnòstic d'HA lleu (5%<FVIII:C): Seqüenciació massiva dels exons i les regions intròniques flanquejants del F8 amb l'aplicació d'un panell de gens relacionats amb les coagulopaties congènites.
- La/es mutació/ns detectades per NGS en estudis d'Hemofília A, per tal d'arribar a un resultat inequívoc, seran recomprovades pel mètode de Sanger analitzant la regió concreta on es troba la mutació.
- En el cas de no identificar cap mutació potencial o definitivament causant de la patologia s'informarà i discutirà amb l'equip mèdic demandant de la prova la possibilitat de realitzar estudis complementaris.
| Codi | Nom de la prova | Es pot demanar per separat? | Es fa sempre? |
|---|---|---|---|
| LRD2224 | Estudi Inversions F8 (intró 22; intró 1) | Si | No |
| LRD2833 | Diag molecular coagulopaties congènites per NGS | Si | No |
Temps de resposta:
20 dies laborables
Informació de l'espècimen
Mostra: Sang total
Tub: Tub EDTA K3 5-10 ml si es tracta d'una mostra de sang
Volum mínim imprescindible: 3 ml
Estabilitat:
- A temperatura ambient: 4 dies
- En refrigeració: 10 dies
Instruccions de transport: Preferiblement a temperatura ambient
Motiu de rebuig: Mostra coagulada i/o incorrectament identificada.
Altres tipus de mostres acceptades:
- DNA purificat, mínim 300 ng (30 ng/µL)
- Mucosa bucal: contactar amb el laboratori per consultar especificacions de recollida de la mostra.
Informació administrativa
Codi BST: LRD2833
Descripció de la prova: Diagnòstic molecular de coagulopaties congènites per NGS: Hemofília A.
Sinònims: Estudi genètic d'HA, estudi molecular de l'Hemofília A, seqüenciació del F8.
Secció: Coagulopaties Congènites
Tarifa BST: Consultar les tarifes actualitzades aquí.
En el full de petició d'estudi molecular s'ha de marcar la casella HEMO. A i omplir les dades fenotípiques de les que es disposi.
Perfils:
No aplica.
Referències
- Peter J Hulick. Next-generation DNA sequencing (NGS): Principles and clinical Applications. Waltham, MA: UpToDate Inc. https://www.uptodate.com
- DNA Sequencing by Capillary Electrophoresis. Applied Biosystems Chemistry Guide. Second Edition.
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99(1):168-174.
- Bagnall RD, Giannelli F, Green PM. Int22h-related inversions causing hemophilia A: a novel insight into their origin and a new more discriminant PCR test for their detection. J Thromb Haemost. 2006;4(3):591-598.
Base de dades de mutacions
- Hemobase: http://www.hemobase.com/
- Human Gene Mutation Database: http://www.hgmd.cf.ac.uk
- EAHAD Coagulation Factor Variant Databases: http://f8-db.eahad.org/